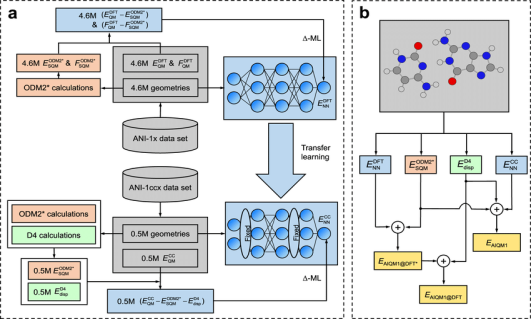
Aromaticity, hydrogen bonding, and metal cofactors are fundamental interactions governing the structure, stability, and function of biomolecular and catalytic systems. Their accurate computational representation remains a major challenge due to the combined influence of electron delocalization, polarization effects, and complex quantum mechanical behavior, particularly in transition-metal environments. Classical molecular mechanics force fields, while computationally efficient, fail to capture these phenomena reliably, motivating the development of quantum mechanical (QM), hybrid QM/MM, and machine-learning (ML) enhanced approaches. This article systematically reviews recent advances in the modelling of aromatic stabilization, hydrogen-bonding dynamics, and metal–ligand coordination using density functional theory (DFT), multi-scale QM/MM simulations, and modern ML potentials. Benchmark systems including aromatic hydrocarbons, hydrogen-bonded clusters, peptide fragments, and biologically relevant metal complexes were analyzed using dispersion-corrected DFT functionals and ML-based force fields trained on high-level QM datasets. Validation metrics such as interaction energies, geometric parameters, aromaticity indices, hydrogen-bond lifetimes, and metal-coordination stability were employed to assess predictive performance. The results demonstrate that modern DFT methods accurately reproduce electronic delocalization and interaction energetics, while QM/MM techniques effectively capture environmental effects in large biomolecular systems. Machine-learning potentials achieve near-QM accuracy at substantially reduced computational cost, showing strong performance for aromatic systems and hydrogen-bond networks, though challenges remain for redox-active metal centers and multi-reference electronic states. Overall, the study highlights that no single modelling strategy is universally optimal. Instead, integrated hybrid frameworks combining QM accuracy, ML efficiency, and classical scalability offer the most promising pathway toward predictive and interpretable simulations. Future progress will depend on metal-inclusive training datasets, physics-informed ML architectures, and improved treatment of polarization and electronic correlation to enable robust modeling across complex chemical space.
| Published in | American Journal of Quantum Chemistry and Molecular Spectroscopy (Volume 10, Issue 1) |
| DOI | 10.11648/j.ajqcms.20261001.12 |
| Page(s) | 15-23 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Aromaticity, Hydrogen Bonding, Metal Cofactors, Quantum Mechanical (QM), Hybrid and Machine-Learning
Method | Strengths | Limitations | Typical Accuracy |
|---|---|---|---|
Classical Force Fields | Fast, scalable, used in MD simulations | Cannot model π-delocalization; no magnetic response; poor for heterocycles | Low for aromaticity (qualitative only) |
DFT (ωB97X-D, M06-2X, B3LYP-D3) | Captures dispersion, π-delocalization, resonance; good for NICS/HOMA | Computationally expensive for large systems; functional-dependent | High (MAE < 1 kcal/mol for resonance energies) |
NICS (Magnetic descriptor) | Excellent for magnetic response; widely used | Overestimates aromaticity in anisotropic fields | High for comparative studies |
HOMA (Geometric descriptor) | Simple, structure-based, useful for large molecules | Geometry alone may not reflect electron currents | Medium–high |
ASE (Energetic descriptor) | Direct measure of stabilization | Requires reference reactions; sensitive to method | High with good reference data |
ML Models (GNNs, equivariant NNs) | Near-QM accuracy with force-field speed; scalable | Needs extensive QM training data; limited interpretability | High for small–medium aromatics |
AIMD | Ab Initio Molecular Dynamics |
AI | Artificial Intelligence |
ANI | Accurate Neural Network Engine for Molecular Energies |
ASE | Aromatic Stabilization Energy |
B3LYP | Becke, 3-parameter, Lee–Yang–Parr functional |
CASPT2 | Complete Active Space with Second-Order Perturbation Theory |
CASSCF | Complete Active Space Self-Consistent Field |
DFT | Density Functional Theory |
DL | Deep Learning |
GAP | Gaussian Approximation Potential |
GNN | Graph Neural Network |
H-bond | Hydrogen Bond |
HOMA | Harmonic Oscillator Model of Aromaticity |
MAE | Mean Absolute Error |
MD | Molecular Dynamics |
ML | Machine Learning |
MP2 | Second-Order Møller–Plesset Perturbation Theory |
NICS | Nucleus-Independent Chemical Shift |
NN | Neural Network |
OPLS | Optimized Potentials for Liquid Simulations |
QM | Quantum Mechanics / Quantum Mechanical |
QM/MM | Quantum Mechanics / Molecular Mechanics |
RMSD | Root Mean Square Deviation |
TD-DFT | Time-Dependent Density Functional Theory |
ωB97X-D | Range-Separated Hybrid Density Functional with Dispersion Correction |
| [1] | Parr, R. G., & Yang, W. (1994). Density-Functional Theory of Atoms and Molecules. Oxford University Press. |
| [2] | Becke, A. D. (1993). Density-functional thermochemistry III. Journal of Chemical Physics, 98, 5648– 5652. |
| [3] | Grimme, S. (2011). DFT with London dispersion corrections. WIREs Computational Molecular Science, 1, 211–228. |
| [4] | Schleyer, P. v. R., et al. (1996). NICS aromaticity probe. JACS, 118, 6317–6318. |
| [5] | Krygowski, T. M., & Cyranski, M. K. (2001). Structural aromaticity. Chemical Reviews, 101, 1385–1420. |
| [6] | Jeffrey, G. A. (1997). An Introduction to Hydrogen Bonding. Oxford University Press. |
| [7] | Scheiner, S. (2011). Hydrogen Bonding: A Theoretical Perspective. Oxford University Press. |
| [8] | Warshel, A., & Levitt, M. (1976). Enzymic reaction electrostatics. Journal of Molecular Biology, 103, 227–249. |
| [9] | Senn, H. M., & Thiel, W. (2009). QM/MM methods. Angewandte Chemie Int. Ed., 48, 1198–1229. |
| [10] | Neese, F. (2012). ORCA program. WIREs Computational Molecular Science, 2, 73–78. |
| [11] | Cramer, C. J., & Truhlar, D. G. (2009). DFT for transition metals. PCCP, 11, 10757–10816. |
| [12] | Jørgensen, W. L., & Tirado-Rives, J. (2005). OPLS-AA force field. Journal of Computational Chemistry, 26, 1689–1700. |
| [13] | Ponder, J. W., & Case, D. A. (2003). Protein force fields. Advances in Protein Chemistry, 66, 27–85. |
| [14] | Smith, J. S., et al. (2017). ANI-1 neural network potential. Chemical Science, 8, 3192–3203. |
| [15] | Schütt, K. T., et al. (2018). SchNet deep learning. Journal of Chemical Physics, 148, 241722. |
| [16] | Behler, J. (2016). Machine learning potentials. Journal of Chemical Physics, 145, 170901. |
| [17] | Liao, M. S., & Siegbahn, P. E. M. (1998). Metalloenzyme electronic structure. Chemical Reviews, 98, 1263–1384. |
| [18] | Friesner, R. A., & Guallar, V. (2005). QM/MM biomolecules. Annual Review of Physical Chemistry, 56, 389–427. |
| [19] | Lin, I.-C., et al. (2012). AMOEBA polarizable force field. JCTC, 8, 2994–3001. |
| [20] | Lan, J., et al. (2023). MACE ML potentials. Nature Communications, 14, 5674. |
| [21] | Grimme, S., et al. (2010). DFT-D3 dispersion correction. Journal of Chemical Physics, 132, 154104. |
| [22] | Zhao, Y., & Truhlar, D. G. (2008). M06 functionals. Theoretical Chemistry Accounts, 120, 215–241. |
| [23] | Chai, J.-D., & Head-Gordon, M. (2008). ωB97X-D functional. Physical Chemistry Chemical Physics, 10, 6615–6620. |
| [24] | Hobza, P., & Šponer, J. (1999). Hydrogen bonding accuracy. Chemical Reviews, 99, 3247–3276. |
| [25] | Berendsen, H. J. C., et al. (1987). Water models. Journal of Physical Chemistry, 91, 6269–6271. |
| [26] | Wang, L.-P., et al. (2014). Force field benchmarking. Journal of Physical Chemistry B, 118, 9956–9972. |
| [27] | Unke, O. T., & Meuwly, M. (2019). PhysNet neural networks. Journal of Chemical Theory and Computation, 15, 3678–3693. |
| [28] | Zhang, L., et al. (2018). DeePMD. Physical Review Letters, 120, 143001. |
| [29] | Bartók, A. P., et al. (2010). Gaussian Approximation Potentials. Physical Review Letters, 104, 136403. |
| [30] | Sousa, S. F., et al. (2017). QM/MM enzyme catalysis. Chemical Reviews, 117, 10851–10909. |
| [31] | Ryde, U. (2016). Transition metals in biology. Coordination Chemistry Reviews, 324, 69–95. |
| [32] | Hohenberg, P., & Kohn, W. (1964). DFT foundations. Physical Review, 136, B864. |
| [33] | Kohn, W., & Sham, L. J. (1965). Self-consistent equations. Physical Review, 140, A1133. |
| [34] | Skylaris, C.-K., et al. (2005). Linear-scaling DFT. Journal of Chemical Physics, 122, 084119. |
| [35] | Cui, Q., & Karplus, M. (2002). QM/MM reaction dynamics. Journal of Physical Chemistry B, 106, 7927–7947. |
| [36] | Grossfield, A., et al. (2018). Force field assessment. Annual Review of Biophysics, 47, 33–52. |
| [37] | Manzhos, S., & Carrington, T. (2021). Neural networks in chemistry. Chemical Reviews, 121, 10187–10217. |
| [38] | Marx, D., & Hutter, J. (2009). Ab Initio Molecular Dynamics. Cambridge University Press. |
| [39] | Cheng, B., et al. (2020). Machine learning free energy. Journal of Chemical Physics, 153, 044118. |
| [40] | Noé, F., et al. (2020). ML for molecular simulation. Chemical Reviews, 120, 11969–12011. |
APA Style
Krishna, R. H. (2026). Integrating Quantum Chemistry and Machine Learning for Accurate Modelling of Aromaticity, Hydrogen Bonding, and Metal Co-Factors. American Journal of Quantum Chemistry and Molecular Spectroscopy, 10(1), 15-23. https://doi.org/10.11648/j.ajqcms.20261001.12
ACS Style
Krishna, R. H. Integrating Quantum Chemistry and Machine Learning for Accurate Modelling of Aromaticity, Hydrogen Bonding, and Metal Co-Factors. Am. J. Quantum Chem. Mol. Spectrosc. 2026, 10(1), 15-23. doi: 10.11648/j.ajqcms.20261001.12
AMA Style
Krishna RH. Integrating Quantum Chemistry and Machine Learning for Accurate Modelling of Aromaticity, Hydrogen Bonding, and Metal Co-Factors. Am J Quantum Chem Mol Spectrosc. 2026;10(1):15-23. doi: 10.11648/j.ajqcms.20261001.12
@article{10.11648/j.ajqcms.20261001.12,
author = {Ravuri Hema Krishna},
title = {Integrating Quantum Chemistry and Machine Learning for Accurate Modelling of Aromaticity, Hydrogen Bonding, and Metal Co-Factors},
journal = {American Journal of Quantum Chemistry and Molecular Spectroscopy},
volume = {10},
number = {1},
pages = {15-23},
doi = {10.11648/j.ajqcms.20261001.12},
url = {https://doi.org/10.11648/j.ajqcms.20261001.12},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajqcms.20261001.12},
abstract = {Aromaticity, hydrogen bonding, and metal cofactors are fundamental interactions governing the structure, stability, and function of biomolecular and catalytic systems. Their accurate computational representation remains a major challenge due to the combined influence of electron delocalization, polarization effects, and complex quantum mechanical behavior, particularly in transition-metal environments. Classical molecular mechanics force fields, while computationally efficient, fail to capture these phenomena reliably, motivating the development of quantum mechanical (QM), hybrid QM/MM, and machine-learning (ML) enhanced approaches. This article systematically reviews recent advances in the modelling of aromatic stabilization, hydrogen-bonding dynamics, and metal–ligand coordination using density functional theory (DFT), multi-scale QM/MM simulations, and modern ML potentials. Benchmark systems including aromatic hydrocarbons, hydrogen-bonded clusters, peptide fragments, and biologically relevant metal complexes were analyzed using dispersion-corrected DFT functionals and ML-based force fields trained on high-level QM datasets. Validation metrics such as interaction energies, geometric parameters, aromaticity indices, hydrogen-bond lifetimes, and metal-coordination stability were employed to assess predictive performance. The results demonstrate that modern DFT methods accurately reproduce electronic delocalization and interaction energetics, while QM/MM techniques effectively capture environmental effects in large biomolecular systems. Machine-learning potentials achieve near-QM accuracy at substantially reduced computational cost, showing strong performance for aromatic systems and hydrogen-bond networks, though challenges remain for redox-active metal centers and multi-reference electronic states. Overall, the study highlights that no single modelling strategy is universally optimal. Instead, integrated hybrid frameworks combining QM accuracy, ML efficiency, and classical scalability offer the most promising pathway toward predictive and interpretable simulations. Future progress will depend on metal-inclusive training datasets, physics-informed ML architectures, and improved treatment of polarization and electronic correlation to enable robust modeling across complex chemical space.},
year = {2026}
}
TY - JOUR T1 - Integrating Quantum Chemistry and Machine Learning for Accurate Modelling of Aromaticity, Hydrogen Bonding, and Metal Co-Factors AU - Ravuri Hema Krishna Y1 - 2026/02/21 PY - 2026 N1 - https://doi.org/10.11648/j.ajqcms.20261001.12 DO - 10.11648/j.ajqcms.20261001.12 T2 - American Journal of Quantum Chemistry and Molecular Spectroscopy JF - American Journal of Quantum Chemistry and Molecular Spectroscopy JO - American Journal of Quantum Chemistry and Molecular Spectroscopy SP - 15 EP - 23 PB - Science Publishing Group SN - 2994-7308 UR - https://doi.org/10.11648/j.ajqcms.20261001.12 AB - Aromaticity, hydrogen bonding, and metal cofactors are fundamental interactions governing the structure, stability, and function of biomolecular and catalytic systems. Their accurate computational representation remains a major challenge due to the combined influence of electron delocalization, polarization effects, and complex quantum mechanical behavior, particularly in transition-metal environments. Classical molecular mechanics force fields, while computationally efficient, fail to capture these phenomena reliably, motivating the development of quantum mechanical (QM), hybrid QM/MM, and machine-learning (ML) enhanced approaches. This article systematically reviews recent advances in the modelling of aromatic stabilization, hydrogen-bonding dynamics, and metal–ligand coordination using density functional theory (DFT), multi-scale QM/MM simulations, and modern ML potentials. Benchmark systems including aromatic hydrocarbons, hydrogen-bonded clusters, peptide fragments, and biologically relevant metal complexes were analyzed using dispersion-corrected DFT functionals and ML-based force fields trained on high-level QM datasets. Validation metrics such as interaction energies, geometric parameters, aromaticity indices, hydrogen-bond lifetimes, and metal-coordination stability were employed to assess predictive performance. The results demonstrate that modern DFT methods accurately reproduce electronic delocalization and interaction energetics, while QM/MM techniques effectively capture environmental effects in large biomolecular systems. Machine-learning potentials achieve near-QM accuracy at substantially reduced computational cost, showing strong performance for aromatic systems and hydrogen-bond networks, though challenges remain for redox-active metal centers and multi-reference electronic states. Overall, the study highlights that no single modelling strategy is universally optimal. Instead, integrated hybrid frameworks combining QM accuracy, ML efficiency, and classical scalability offer the most promising pathway toward predictive and interpretable simulations. Future progress will depend on metal-inclusive training datasets, physics-informed ML architectures, and improved treatment of polarization and electronic correlation to enable robust modeling across complex chemical space. VL - 10 IS - 1 ER -
Department of Chemistry, Amrita Sai Institute of Science and Technology, Vijayawada, India
Figure 1. Accurate Modelling of Aromaticity, Hydrogen Bonding, and Metal Cofactors.
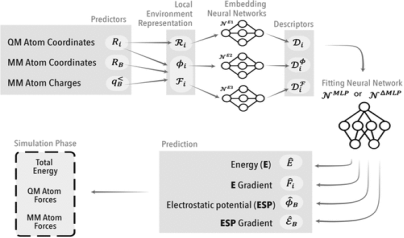
Figure 2. Overview of computational methodologies used in this study.
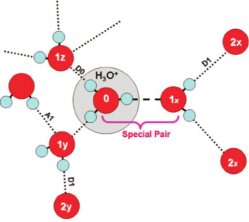
Figure 3. Representative benchmark systems used to evaluate the modelling of aromaticity, hydrogen bonding, and metal cofactors.
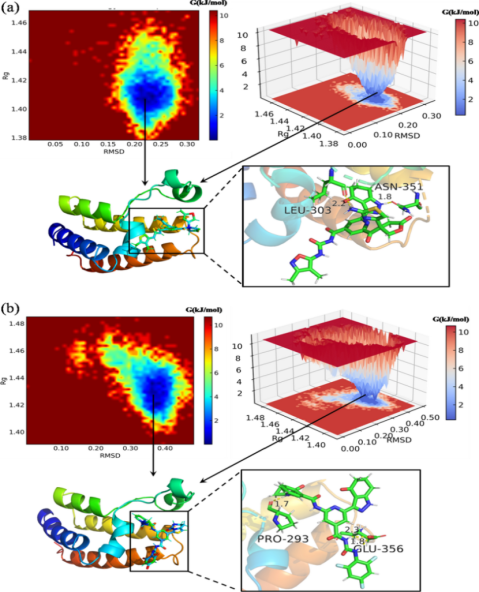
Figure 4. Key validation metrics used for accuracy assessment.
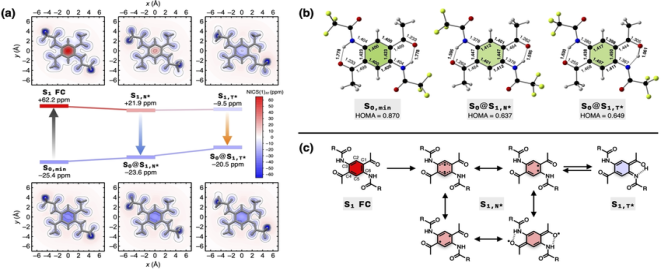
Figure 5. Overview of aromaticity modelling approaches.
Information

